
【新智元导读】昨日Nature封面论文:哈佛大学研究者借助机器学习算法,利用“废弃”数据成功预测新材料的合成,引发学界激论:人工智能真能加速发现神奇新材料吗?该研究所用的“计算材料学”结合计算机模型和机器学习,是对传统研究方法的革新。计算机科学和人工智能的影响已经拓展到越来越多的领域,机器学习或将改变未来科研方式。
发现一种新的材料是非常艰难的过程,通常要经历无数次失败,偶尔在机缘巧合之下取得成果,还要费劲功夫反向检测这种新材料的性质。但有一批材料科学家转换思路,使用计算机模型和机器学习算法生成海量假想的材料,建立数据库,从中筛选出值得合成的材料,再通过检索这些材料可能拥有的性质进行具体应用测试,比如将这种材料用作导体表现如何、用作绝缘体性能又如何、这种材料是否具有磁性、那种材料的抗压力是多少。
2016年5月5日,Nature 将一篇机器学习算法改变材料发现方式的论文放上封面,并提出“从失败中学习”:哈佛大学研究者利用机器学习算法,用失败或不成功的实验数据预测了新材料的合成,并且在实验中机器学习模型预测的准确率超过了经验丰富的化学家,这意味着机器学习将改变传统材料发现方式,发明新材料的可能性也大幅提高。
使用计算机模型和机器学习算法的好处在于,失败的实验数据也能用作下一轮的输入,继而不断完善算法。伦敦帝国学院研究副院长、材料科学家 Neil Alford 以观察者身份发表评论,这种做法代表了实验科学和理论科学的真正融合。
加州大学伯克利分校的材料科学家 Gerbrand Ceder 在接受 Nature 记者采访时说,使用机器学习算法有望大幅提高新材料发现的速度和效率。Ceder 是最早开始使用计算模型和机器学习生成假想材料的科学家之一,他以化合物磷酸铁锂为例:磷酸铁锂最初于 20 世纪 30 年代被合成,但当时世人并不认为这种材料会有多大用途,直到 1996 年科学家发现磷酸铁锂大有取代现有锂离子电池的可能。
Haverford College和Purdue University的研究者采用计算材料科学思路,使用“失败”数据,成功完成了这篇被选为本期 Nature 封面的论文。
有了机器学习,再也不怕失败了
. 论文标题:Machine-learning-assisted materials discovery using failed experiments
. 作者:Paul Raccuglia、Katherine C. Elbert、Philip D. F. Adler、Casey Falk、Malia B. Wenny、Aurelio Mollo、Matthias Zeller、Sorelle A. Friedler、Joshua Schrier、Alexander J. Norquist
. 来源:Nature 533, 73?76 (05 May 2016) doi:10.1038/nature17439
使用失败实验在机器学习辅助下进行材料发现(摘译)
对诸如有机模板合成的金属氧化物、金属有机骨架(MOF)和有机卤化钙钛矿等无机-有机杂化材料的研究已经持续了数十年。水热法和(非水)溶剂热合成已经产生了数千种新材料,这些新材料几乎包含了元素周期表中的所有元素。然而,我们仍未充分理解这些化合物的形成过程,对新化合物的开发主要依靠试探性合成。在Materials Genome Initiative的推动下,计算机模拟和数据驱动的方法成为对实验试错方法的替代选择。三个主要的策略是:基于模拟来预测材料的电荷迁移率、光生伏打性质、气体吸附能力和锂离子嵌入等物理性质,从而确定那些有前景的合成对象。通过整合高通量合成与测量工具,从大规模实验数据中确定材料的结构-性质关系。基于诸如沸石结构分类和气体吸附性能等相似的晶体结构,对材料进行聚类。
在这里,我们展示了用反应数据训练机器学习算法,继而预测模板合成的钒亚硒酸盐结晶过程的反应结果。我们使用未发表的“黑暗”反应信息,这些反应信息来自那些失败或未成功的水热合成实验。我们从实验室的笔记本档案中收集了这些信息,并运用化学信息学技术为笔记本中的原始数据添加了理化性质描述。我们用由此产生的数据训练机器学习模型预测反应能否成功。当使用先前未经测试的、市场有售的有机砌块进行水热合成实验时,我们的机器学习模型获得了比传统人类策略更好的效果,并成功预测了有机模板合成的无机物的形成条件,成功率达 89%。对机器学习模型进行反演后,可以揭示出关于成功产物形成条件的崭新假设。
实验中机器学习模型反馈机制示意图
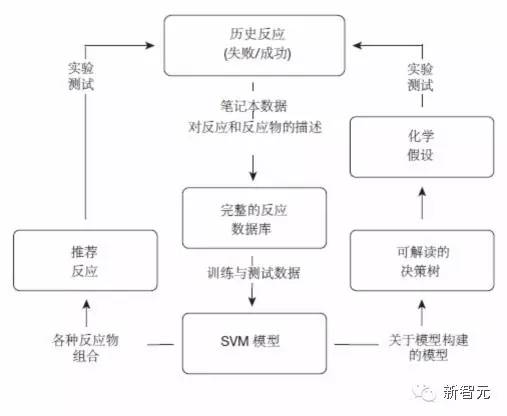
图1|“黑暗”反应的反馈机制示意图。使用从历史反应数据中产生的机器学习模型推荐可供执行的新反应,并产生关于结晶过程的假设,这些假设可以被人类解读。另,SVM 是支持向量机的缩写。来源:Nature 533, 73?76
机器学习模型超越传统人类策略
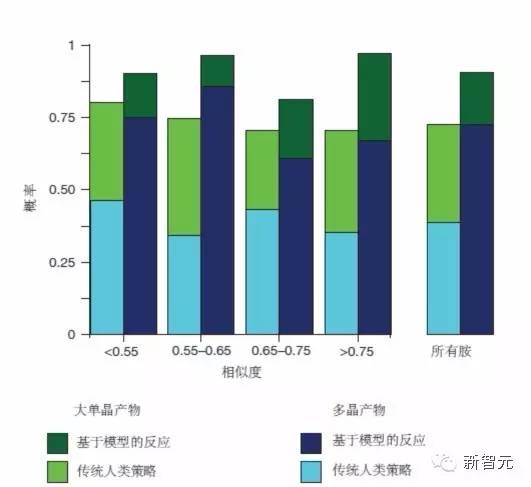
图2|关于模板合成的钒亚硒酸盐晶体形成的实验结果比较,以胺相似度为横轴。深色条表示机器学习模型的预测,浅色条表示传统的人类策略。产生了多晶和大单晶产物的反应分别显示为蓝色和绿色。纵轴显示了反应出现所指示的结果的概率。机器学习模型比人类策略更成功地预测了晶体形成的条件,无论用模板合成的胺数据库中已知实例时所具有的系统相似性如何。来源:Nature 533, 73?76
支持向量决策树
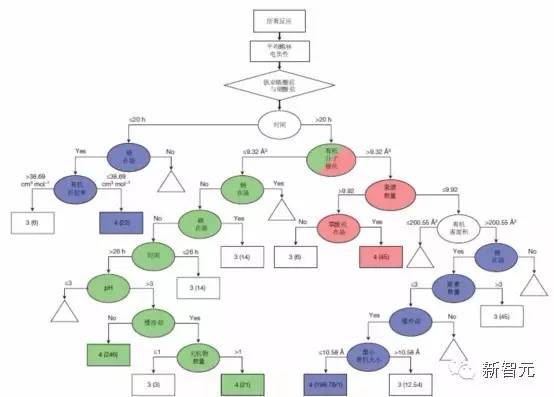
图3|从 SVM 中得到的决策树。椭圆表示决策节点,矩形代表反应结果容器,三角形代表被切除的子树。箭头上的数字对应于决策属性的测试值。每个反应结果容器(矩形)对应一个特定的反应结果值(“3”或“4”,如图所示)。括号中的数字是正确地分配给该容器的反应的数量(任何被错误分类的反应都用正斜杠标识)。分数值表示反应具有不确定的结果,这是由决策树的较高位置的属性值缺失导致的。那些包含了大多数成功反应的容器被分为三个不同的组(分别用绿色,蓝色和红色阴影标示)。每个彩色子树定义了一组有助于单晶形成的特定反应参数。通过审查这些条件,可以得出相应的化学假设,这些假设分别对应于低、中和高极化胺。来源:Nature 533, 73?76
算法生成的假设及其化学三维结构模型
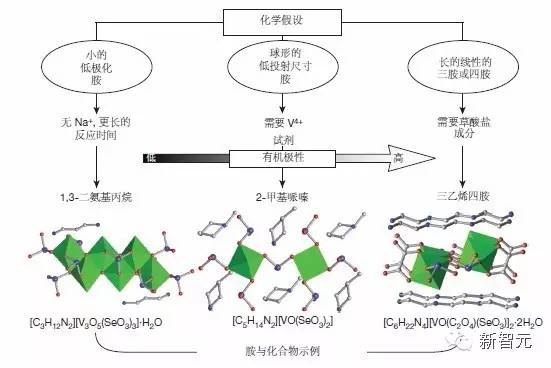
图4|对从模型中产生的三个假设及每个假设结构的图示。单晶形成所需的实验条件很大程度上取决于胺属性。小的、低极化的胺需要不存在与之竞争的 Na+ 离子,也需要较长的反应时间,以避免无机砌块沉淀。球形、低投影尺寸的胺则需要包含 VOSO4 等试剂的 V4+ ,因为它们不能直接从常见的 V5+ 前体中产生 V4+。长的三胺和四胺要求草酸盐反应物,以改变无机次级砌块的电荷密度。这三个假设分别对应于图 3 中的绿色、蓝色和红色子树。
我们的机器学习方法使我们能够利用包含历史反应的化学信息,并阐明支配反应结果的因素。机器学习模型对先前未经测试的有机胺的预测准确率,超过了依靠多年来形成的化学直觉所实现的准确率。此外,我们的方法以可验证的假设形式揭示了支配反应结果的化学原理,它能更成功地制造新化合物,也能产生有用的化学信息,这代表了试探性反应的革新性进步。
AI 真能发现神奇材料吗?
计算材料科学还是一门新兴的学科,其主要推动着就是上文提到的加州大学伯克利分校的材料科学家 Gerbrand Ceder 。受人类基因组计划的启发,Ceder 想到了使用高通量数据驱动的方法进行材料发现。Ceder 认为,人类基因组本身并非能作为疾病治疗的方案,但却可以为医学提供研发疾病治疗方案的海量基本定量数据??材料科学是不是也能借鉴遗传科学的方法,用“材料基因组”(该词为 Ceder 所创)编码各种化合物呢,就像 DNA 碱基对编码蛋白质等各种生物材料一样?
2003 年,Ceder 研究组创建了一个量子力学计算数据库,用于预测金属合金最有可能形成的晶体结构,因为这是发明新材料的基础。在过去,即使使用用超级计算机也需要通过多次反复长期大量试错找到合金的基态。但在 Ceder 研究组 2003 年发表的一篇论文中,他们描述了一种捷径:研究人员首先计算出一些常见二元合金晶体结构的能量,建立小型数据库,然后设计了一种机器学习算法,这种算法可以从上述数据库中提取模式,继而预测出新合金基态的可能值。结果表明,Ceder 研究组设计的这种机器学习算法表现良好,大大缩减了计算时间。
2006年,Ceder 在 MIT 开始了 Materials Genome Project,用改进后的机器学习算法预测能用作电动车电池的锂材料。2010年,该计划的数据库里已经包含了2万种计算机预测的化合物。另一方面,Ceder 研究组成员 Stefano Curtarolo 在 2006 年去了杜克大学并在那里建立了自己的实验室??Center for Materials Genomics,专门研究金属合金,Curtarolo 研究组与其他两家研究机构合作,逐渐改进 2003 年的机器学习算法并拓展数据库,构建了 AFLOW 系统,能计算已知的晶体结构并且自动预测新的晶体结构。
2011年6月,白宫宣布斥资几亿美元进行 Materials Genome Initiative(MGI),由此开始计算材料科学这门学科成为主流。如今,除了 Ceder 的 Materials Project,还有原 Ceder 研究组成员、现杜克大学材料科学家 Stefano Curtarolo 的数据库 AFLOWlib,以及西北大学材料研究者 Chris Wolverton 在 Ceder 思路启发下,用自己研发的算法和模型建立的数据库 Open Quantum Materials Database(OQMD)。
这3大数据库都含有从材料科学界广泛使用的无机晶体结构数据库中提取的5万种材料,这些都是曾经被制造出来的固体,但其导电性和磁性尚未被彻底研究。其不同之处在于:Ceder 的 Materials Project 侧重沸石、锂电池相关以及金属有机骨架结构材料,并以较高的标准衡量是否将计算机预测的材料纳入数据库;Curtarolo 的 AFLOWlib 是最大的数据库,包含 100 多万种材料和几十万种假想材料,但相应的里面也不乏只能存在一瞬间的材料;Wolverton 的 OQMD 有大约 40 万种假想材料,其中钙钛矿相关的尤其丰富,此外正如名字中 Open 那样,用户可以下载整个数据库。
目前这3大数据库都在用各自的方法不断补充数据、完善算法,但离理想还有很大距离。当前的机器学习算法相对擅长预测某种晶体是否稳定,但在预测吸光性和导电性时则会出现很大误差。不过,Materials Project 已经发现了几种有望超越现有锂离子电池阴极材料性能的材料,以及有可能提高太阳能电池能量转化率的金属氧化物。都柏林三一学院的研究人员使用 AFLOWlib 预测了 20 种可用于制作传感器或计算机存储器的磁性材料,并且成功合成了其中的两种,同时经实验证明其磁性与预测非常接近,相关论文已经在 Nature 发表。
欧洲也有类似的计算材料计划:由瑞士洛桑联邦理工学院(EPFL)为首的一批计算材料科研机构共同组建了 MARVEL,EPFL 的材料科学家 Nicola Marzari 是该项目的负责人。Marzari 正在使用新的计算平台制作一个叫做 Materials Cloud 的数据库,主要用于搜索石墨等由一层原子或分子组成的“二维”材料,这类材料可以在纳米电子、生物医学设备领域得到广泛应用。Marzari 的 Materials Cloud预 计今年晚些时候启动,学界也对此表示了普遍的关注。据 Mzrzari 预计,到 Materials Cloud 开放时,系统将会得出大约 1500 种有望进入试验阶段的二维材料结构。
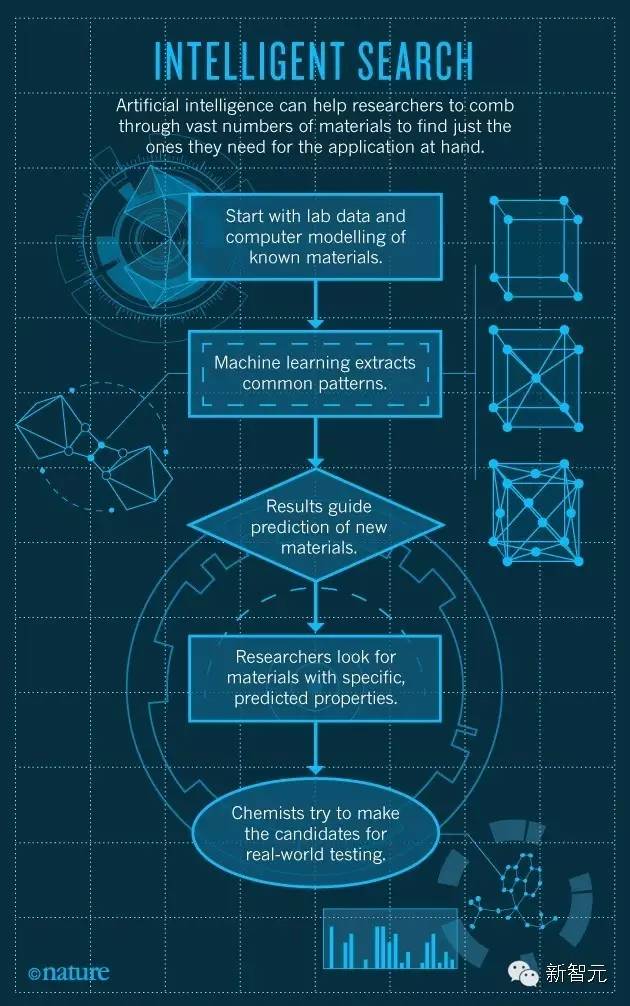
人工智能帮助科学家发现新材料。来源:Nature
不过,计算材料的发现也不全是好的结果:EPFL 中心的计算化学家 Berend Smit 及其研究组筛选了计算机预测的 65 万种材料后得出结论,当前用于存储甲烷的材料基本已经是最好的了,纵使得到改善,存储效率也只能微量提升,这说明美国寄希望于重大技术突破(如使用纳米多孔材料存储甲烷)而设定的能源目标很可能是不现实的。
目前,Ceder 和 Curtarolo 都在努力开发更好的机器学习算法,从已知化合物合成过程中提取规律。Marzari 告诉 Nature 记者,材料科学已经从手工时代进入了产业化阶段,虽然现在市面上还没有计算材料得到应用,但他相信十年后不仅会有,而且可能会有很多。
不过,就连支持使用计算机和机器学习生成假想材料的科学家也指出,要从假想材料到现实落地还有很长一段距离。首先,现有数据库所含有的材料数据本身就不多,连现有已知材料都没有收录完全,更被说计算机生成的材料了。其次,这种用数据驱动的发现方法并不适用于所有的材料(目前算法只能预测完美晶体)。再者,即使计算机生成了一种极有前景的材料,要在实验室里将其合成、制为实物也仍然可能需要花费很长时间。Ceder 对 Nature 记者说,计算机随时都在生成有趣的新材料,但有时候半年多时间都无法在实验室里将其制造出来。换句话说,在理论上合成一种材料相对简单,但要在实验室里把它做出来很难。
但是,材料科学家对于发现新的化合物充满信心,他们相信还有数不清的新材料有待合成,而这些新材料将对电子工业、能源产业、机器人产业、健康医疗和交通运输带来巨大改变。


 发表于 2015-11-9 11:08:32
发表于 2015-11-9 11:08:32

 楼主
楼主